确定
先天性睑裂狭小综合征与FOXL2基因
发布于 2022-03-07 来源:复禾健康
概述
先天性睑裂狭小综合征
(BPES)是一种罕见的常染色显性
遗传病
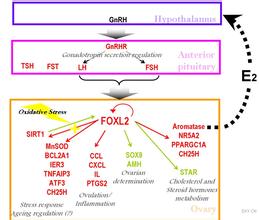
。BPES分为两型,研究表明FOXL2基因是BPES的致病基因,在BPESⅠ型和Ⅱ型患者中均存在FOXL2基因突变。中外许多研究者对BPES家系或者散发病例的FOXL2基因突变进行了研究。
步骤/方法
01
第一步:BPES临床上以睑裂狭小,上睑下垂,逆向内眦赘皮,内眦远距为主要征象。偶有散发病例。部分患者伴有智力低下,生长迟缓,心房或室间隔缺损等。VonAmmon于1841年最先描述此病,并指出有遗传性。
02
第二步:BPES分为两型:Ⅰ型,由父亲传代,女性患者因卵巢功能早衰(POF)而不育,男性生育功能正常。外显完全,外显率为100%,女性患者有不孕症,原发闭经和提前绝经,小子宫及卵巢萎缩。Ⅱ型,父亲、母亲传代机会均等,男女患者均只累计眼部而可以生育,不完全外显,外显率约为96.5%。
03
第三步:FOXL2基因显然与另一基因SOX9保持着排斥关系。当一种基因启动,另一种则自动关闭。SOX9基因通常只在男性体内活动,当男性的SOX9一旦被开启,FOXL2的活动就遭抑制,并进而终身停顿。这种情况在女性体内刚好相反,FOXL2会最先被启动。学界普遍了解FOXL2对女性维持女儿身与卵巢的成长十分重要,然而科学家并不预期卵巢中的排卵细胞会被SOX9基因吸收,进而发挥男性生育功能。
注意事项
研究人员认为,该发现离人体应用还有一段很长的路要走,然而这必然带来变性治疗的变革,甚至可能开启非手术变性治疗的先河。到时,变性人将无须终生用药,只需接受短期的基因疗法就行了。
温馨提示:文章内容仅供参考,如果您有就诊需求,可以将病症信息提交给我们,以便我们能及时与您联系!
预约就诊
相关经验